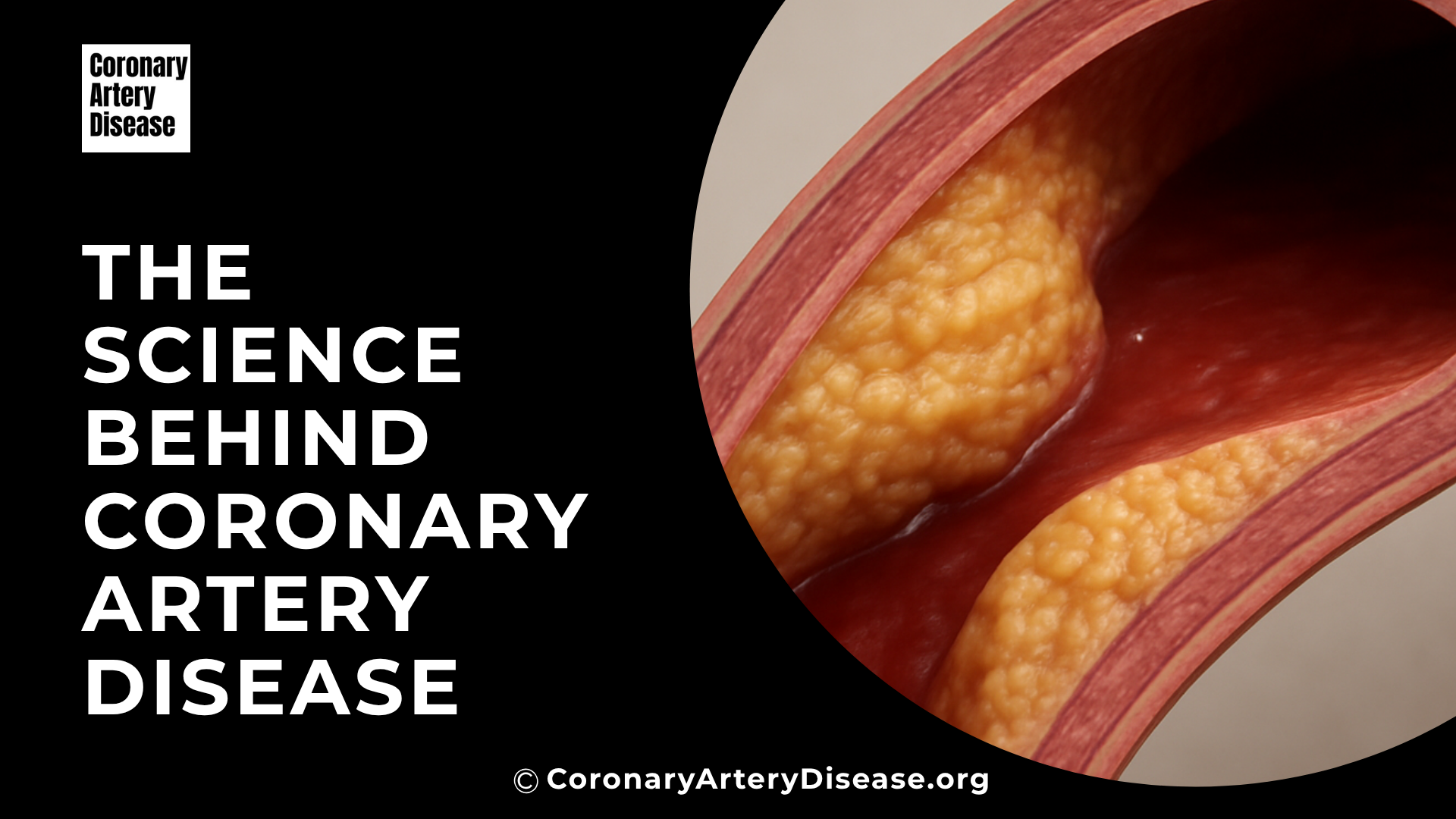
Everything you need to know about coronary artery disease and atherosclerosis
Our mission is to lay the groundwork for a new era of discovery, enabling medical students and cardiovascular researchers to transform our understanding of coronary artery disease, the world’s foremost cause of mortality.